This page provides introductory and background information on various glycoengineering strategies including metabolic glycoengineering (MGE), a specialty of our lab’s research (Section 1.1); we next describe our team’s efforts to improve monosaccharide analogs used in MGE (Section 1.2); and finally, we describe a multifaceted glycoengineering platform that progressively combines complementary strategies to design improved therapeutic proteins (Section 1.3).
1.1 Introduction and Background
1.1.1 Definition of Glycoengineering. Glycoengineering is a broad term that refers to the manipulation of glycans, which are sugar structures attached to proteins, lipids, nucleic acids, and extracellular matrix components:
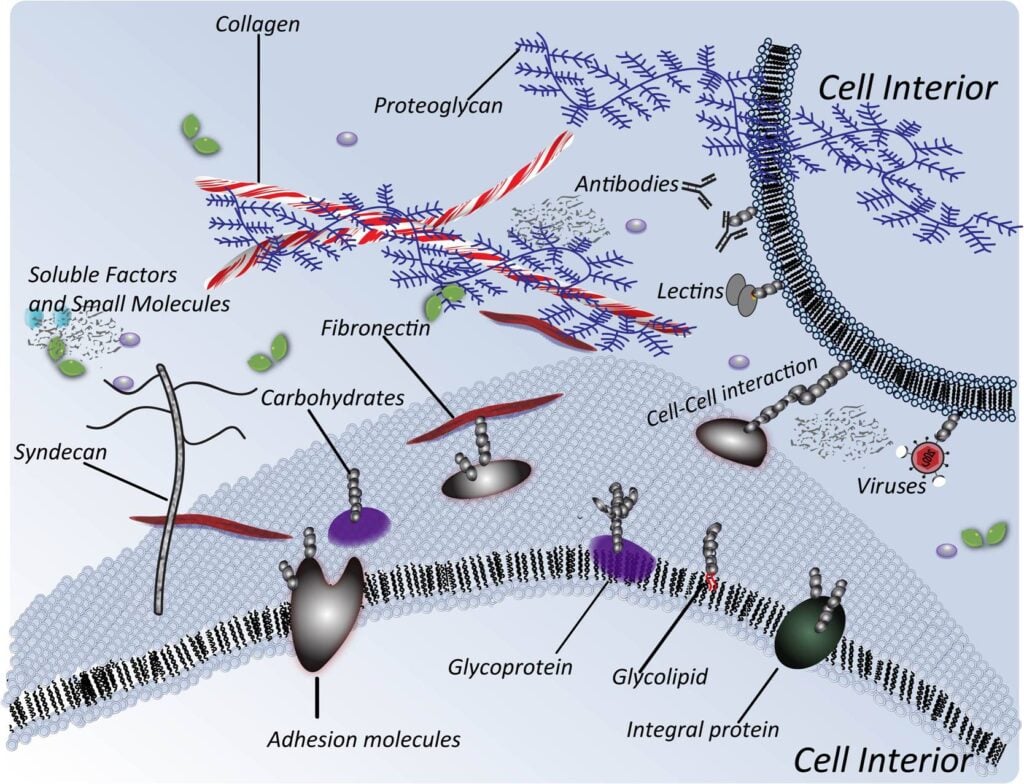
Here (below) we focus on two glycoengineering techniques that have biomedical potential, specifically “building in” sites of glycosylation (Section 1.1.2) and metabolic glycoengineering (MGE, Section 1.1.3).
1.1.2 Erythropoietin (EPO), a Pioneering Therapeutic Protein Improved with “Built in” N-Glycans. Virtually all regulatory proteins found in the body including hormones, cytokines, antibodies, and cell surface receptors, are glycosylated. The ubiquitous nature of N-glycosylation in particular supports the idea that, at least in some cases, the deliberate “building in” of additional glycans can modulate and improve protein function. A pioneering example of a therapeutic protein improved by this approach is provided by darbepoetin alfa, a form of EPO that has five N-glycans compared to the native protein, which has three.
, which has two \"built in\" N-glycans.")
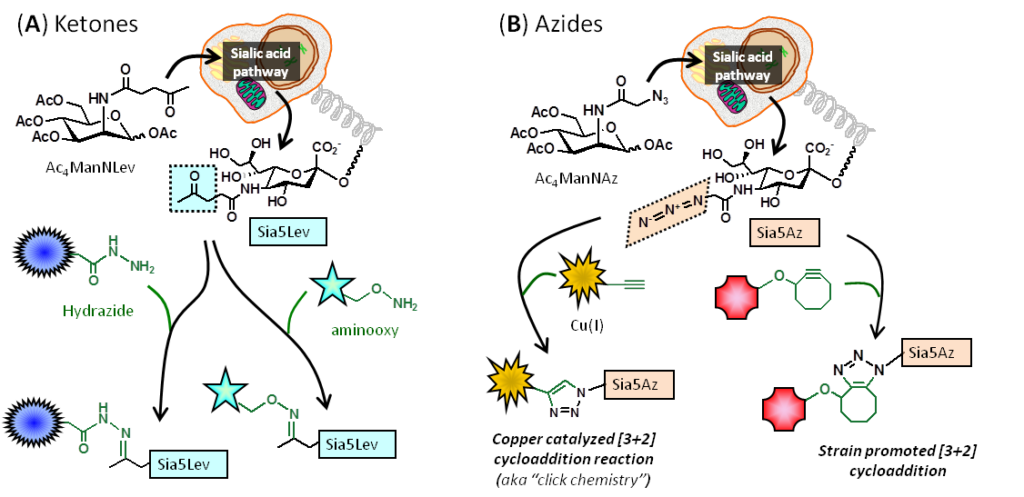
1.1.3 Introduction to Metabolic Glycoengineering (MGE). Metabolic engineering refers to a technique pioneered in the 1990s in living cells and animals largely by Werner Reutter‘s and Carolyn Bertozzi‘s research groups. This technology facilitated the installation of various bioorthogonal, non-natural chemical functional groups onto the cell surface include ketones and azides:
1.2 Improving Metabolic Glycoengineering (MGE) through Monosaccharide Analog Design
1.2.1 Pitfalls – and Solutions – for ‘Early Generation’ MGE Analogs. The use of MGE analogs, e.g., azido-modified ManNAc used to install azido groups into cell surface sialic acids (Figure 1.1.3B), suffered from the inefficiency of N-acyl modified hexosamine analogs, with are not able to enter cells through monosaccharide transporters. A solution to this problem involved adding ester-linked acetate groups to the MGE analog, resulting in increased lipophilicity and non-specific membrane diffusion; for example, Ac4ManNAz is up to three orders of magnitude more efficient than ManNAz:
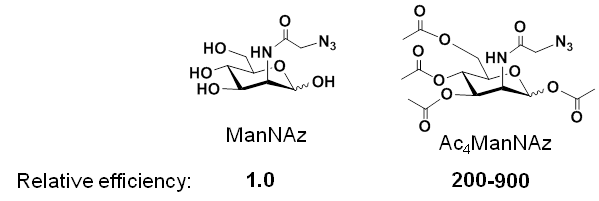
Once a peracetylated analog such as Ac4ManNAz enters a cell, non-specific intracellular esterases remove the acetate groups, allowing the core ManNAz to enter the sialic acid biosynthetic pathway.
Based on the mechanism of peracetylated analogs, i.e., their increased lipophilicity increases non-specific membrane diffusion into a cell, our team reasoned that attaching longer short chain fatty acids to the analog would further increase membrane diffusion and analog efficiency. Accordingly, we created propyl- and butyl-modified analogs and found further increases in efficiency:
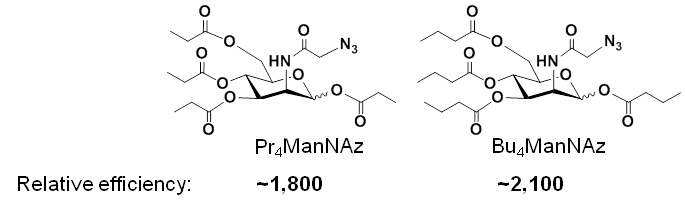
A drawback of Ac4ManNAz, Pr4ManNAz, and Bu4ManNAz is increasing cytotoxicity of each analog that hinders the use of the analogs in MGE applications. Our team solved this problem by omitting the ester-linked SCFA from the C6-OH position; the resulting analog (1,3,4-O-Bu3ManNAz, Figure 1.2.1(C) below) not only was less toxic than Bu4ManNAz but also more efficient than Bu4ManNAz. Interestingly, arrangement of three ester-linked butyrate groups on the core hexosamine leaving the C1-OH position unmodified endowed the analog (e.g., 3,4,6-O-Bu3ManNAz) with dramatically different biological activity:
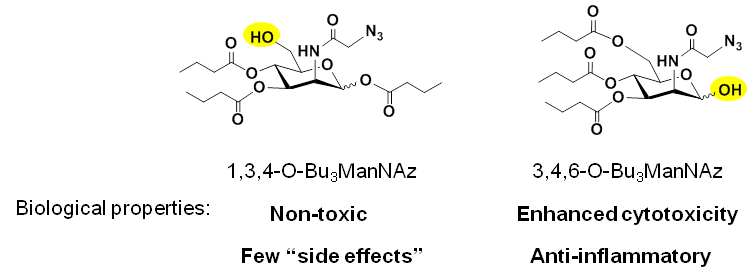
The discovery that the “1,34,” vs “3,4,6” stereochemistry of hexosamine analogs tuned their bioactivity opened the door for distinct applications for each set of compounds; selected applications are described below for the “1,3,4” (Section 1.2.2) and “3,4,6” (Section 1.2.3) analogs.
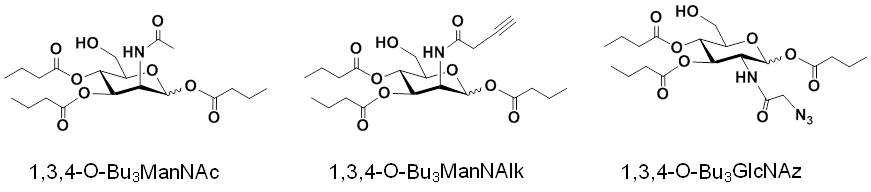
1.2.2 Properties, Benefits, and Applications of “1,3,4” Hexosamine Analogs. The non-toxic, ‘high flux’ properties of 1,3,4-O-Bu3ManNAz apply broadly to hexosamines with different N-acyl groups (e.g., the natural N-acetyl group, as 1,3,4-O-Bu3ManNAc [Figure 1.2.2(A)] or alkynes, as 1,3,4-O-Bu3ManNAlk) as well as to other hexosamines (e.g., GlcNAc, as 1,3,4-O-Bu3GlcNAz):
1.3 A Multifaceted Production Platform for Improving Therapeutic Proteins
1.3.1 Improving Therapeutic Proteins through Complementary Glycoengineering Strategies. As described above, the darbepoetin alfa form of EPO is a pioneering example of a successfully glycoengineered drug (Figure 1.1.2). Only one mode of glycoengineering was used, however, which was the addition of N-glycan sites. By contrast, we have developed a multidimensional platform for glycan (and protein) engineering therapeutic proteins that combines multiple strategies to improve monoclonal antibodies (mAbs, Section 1.3.2, below) and a therapeutic enzyme (Section 1.3.3).
1.3.2 Enhancing mAb Glycosylation and Production.
1.3.2 Benefits of 1,3,4-O-Bu3ManNAc, a “high flux” analog that enhances sialylation in sialyltransferase-overexpressing cells. We demonstrated the benefits of combining complementary glycoengineering strategies to improve therapeutic proteins by producing mAbs in Chinese hamster ovary (CHO) cells that simultaneously over-expressed glycogenes and were supplemented with 1,3,4-O-Bu3ManNAc. In particular, ST6GAL1, a human α-2,6-sialyltransferase, was overexpressed to increase (and “humanize”) sialylation of recombinant mAbs produced in the host CHO cells (Yin et al, 2015). In concert, the cells were supplemented with 1,3,4-O-Bu3ManNAc to increase flux through the sialic acid biosynthetic pathway to meet the increased metabolic demands for this sugar (Yin et al, 2017). Interestingly, the amount of mAbs produced in analog-supplemented cells increased, which was attributed to the intracellular liberation of n-butryate from 1,3,4-O-Bu3ManNAc (Wang et al, 2019). Importantly, unlike previous attempts to use n-butryate to increase the production of therapeutic proteins where glycan quality was compromised (e.g., sialylation was decreased), supplying n-butyrate as part of 1,3,4-O-Bu3ManNAc did not decrease glycan quality. Overall, the combined use of the glycoengineered CHO cells as production hosts and supplementation with the analog resulted in combined benefits making our strategy attractive for biopharmaceutical manufacturing. Moreover, our production platform proved to be versatile, as evidenced by similar benefits observed for ENPP1, a therapeutic enzyme (described below, in Section 1.3.3).
1.3.2.2 Combining 1,3,4-O-Bu3ManNAc supplementation with “built-in” glycans. Based on consolidating evidence that the pharmacological properties and bioactivity of monoclonal antibodies (mAbs) can be modulated by non-canonical Fab glycans, we added N-glycans to two mAbs (F5111 and 602, Figure 1.3.2.2, Ludwig et al, 2022).
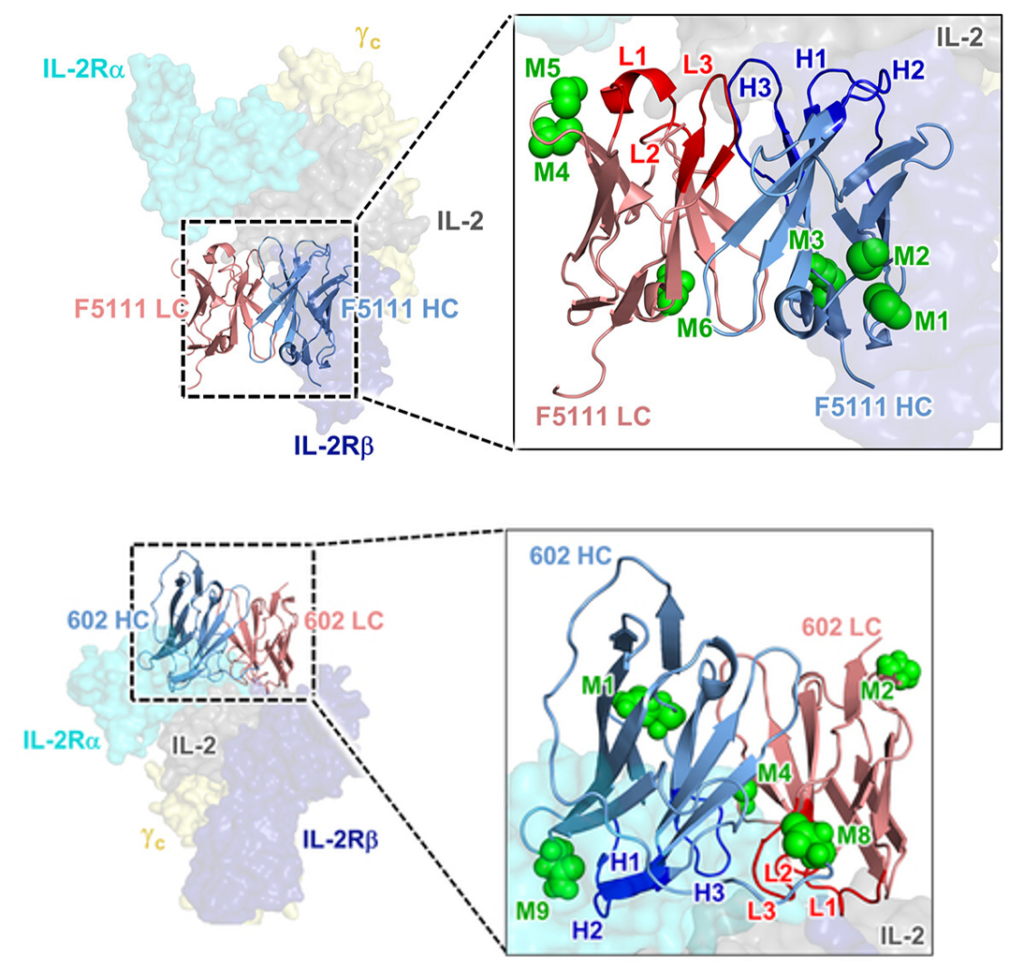
The glycovariants we created exhibited modified activities compared to the parent antibody, showing the potential of our glycoengineering strategy to modulate biological function of antibodies involved in multi-component receptor systems. Finally, when coupled with a high-flux sialic acid precursor, a glycovariant with two installed glycosylation sites demonstrated superior in vivo half-life. Collectively, these findings validate a versatile glycoengineering strategy that introduces atypical glycosylation into therapeutic antibodies in order to improve their efficacy and, in certain instances, modulate their activity early in the drug development process.
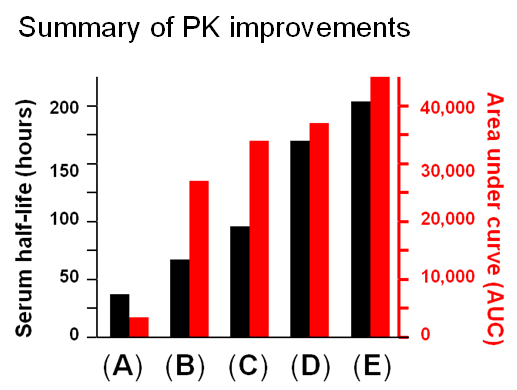
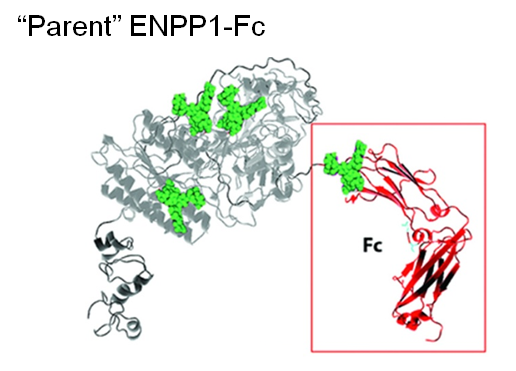
1.3.3 Improving ENPP1 through Protein and Glcyoengineering. ENPP1 is ectonucleotide pyrophoshatase/phosphodiesterase 1, a blood enzyme whose deficiency results in generalized arterial calcification of infancy (GACI), a potentially lethal congenital disease. Wild-type ENPP1 has a short serum half-life of about 5 hours when used for enzyme replacement therapy (ERT), necessitating thrice a day dosing in a mouse model of GACI. To improved the pharmacokinetic (PK) and pharmacodynamic (PD) properties of ENPP1, the Braddock group fused an IgG Fc domain to this enzyme, improving serum half-life to about 37 hours (Albright et al, 2016 and Figure 1.3.3(A)).
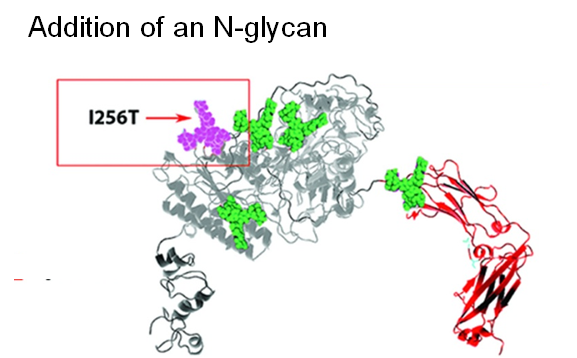
Next, by taking a glycoengineering approach, a fifth N-glycan was added to ENPP1-Fc, which almost doubled the serum half-life to 67 hours while AUC, a surrogate measure of bioavailability increased dramatically from 3,400 to 27,000 units (Figure 1.3.3B).
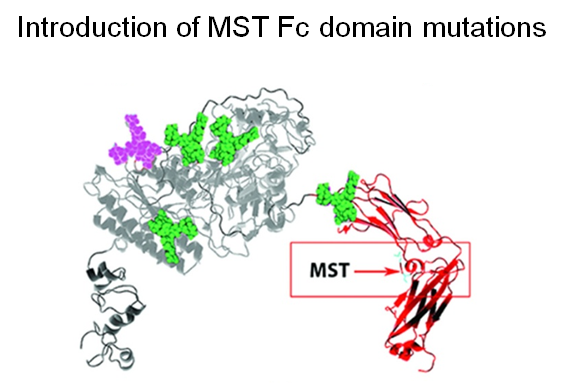
A further improvement to the PK and PD properties of ENPP1-Fc was next achieved by another protein engineering strategy through MST mutations that increase affinity for the neonatal Fc receptor (Figure 1.3.3C).
Finally, two approaches were taken to increase sialylation. First, glycoengineered “MST” ENPP1-Fc was produced in α-2,6-sialyltransferase overexpressing CHO cells (Figure 1.3.3D) followed by supplementation with 1,3,4-O-Bu3ManNAc, a high flux precursor that feeds metabolic flux into the sialic acid biosynthetic pathway (Figure 1.3.3.E).
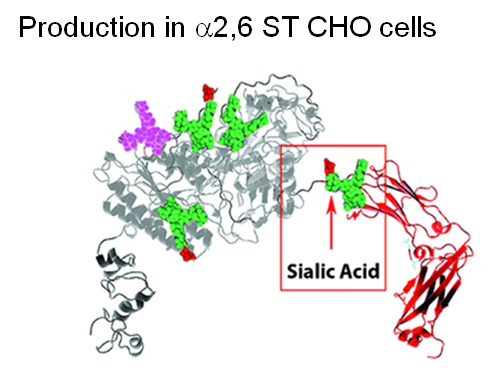
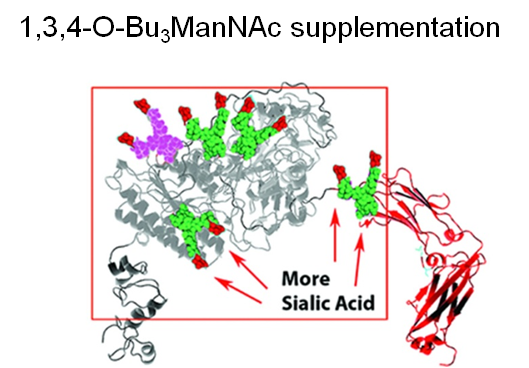
Overall, the step by step improvements outlined in Figure 1.3.3, Panels A through E (as summarized quantitatively in Panel F) comprise an integrated glycoengineering platform capable of dramatically improving the PK and PD properties, and hence the efficacy, of therapeutic proteins.